A GAME CHANGER FOR GENE THERAPY FOR RETT SYNDROME.

Background.
As noted in previous articles now on the RSAA website, gene therapy for Rett Syndrome represents a potential cure. Other potential strategies are being explored but gene therapy probably represents the strategy most likely to reach the clinic first. Many of these strategies use an adeno-associated virus (AAV) to deliver the MeCP2 gene to neurons in the brain.
Limitations of gene therapy.
Several issues still need to be overcome:
- AAV is a small virus so there is a limit to the size of any gene which can be inserted into the AAV genome while other elements which normally control MeCP2 expression cannot be inserted and
- because Rett is an X-chromosome linked syndrome, only 50% of neurons have a defective MeCP2 gene. The AAV-MeCP2 cannot discriminate between neurons that contain a functional or mutant MeCP2 gene.
This raises the probability that MeCP2 will be over-expressed in neurons which contain a functional MeCP2 gene, in turn leading to the possibility of MeCP2 duplication syndrome.
Avoidance of MeCP2 duplication syndrome and MeCP2 over expression.
Years of research have failed to overcome the dilemma of MeCP2 over-expression leading to MeCP2 duplication syndrome versus MeCP2 under-expression resulting in no change in the behaviour of mice with Rett Syndrome. In a recent landmark study, a strategy to control the level of MeCP2 expression and thus avoid MeCP2 duplication syndrome was recently described in a paper from Professor Steven Gray’s laboratory in the University of Texas, Dallas (Sinnett SE et al, 2021. Engineered microRNA-based regulatory element permits safe high-dose miniMeCP2 gene therapy in Rett mice. Brain May 5;awab182. doi: 10.1093/brain/awab182).
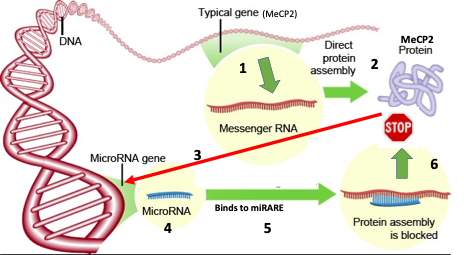
MicroRNA (miRNA) is another type of RNA produced in a cell and recognises messenger RNA (mRNA-the intermediate template molecule that results in production of a protein). The miRNA binds to mRNA resulting in loss of expression of the protein. Professor Gray’s team hypothesized that it should be possible to find a miRNA that can bind to MeCP2-specific mRNA and thus inhibit MeCP2 expression.
Professor Gray and his team identified several miRNA molecules which were expressed in cells as a by-product of MeCP2 over-expression. These miRNA molecules recognise an independent target which was termed a miRNA-Responsive Auto-Regulatory Element (miRARE), “a built-in inhibitory element responsive to MeCP2 overexpression”.
The use of a miniMeCP2 gene freed up space in the recombinant AAV-MeCP2 genome to permit the inclusion of the miRARE element. Thus, after delivery of the AAV-MeCP2-miRARE to neurons in mice, only neurons which contained the defective MeCP2 protein were shown to express the truncated (functional) MeCP2 protein encoded by the miniMeCP2 gene and resulted in significant improvement in the Rett severity scores. The principle of the process is explained in Figure 1.
FIGURE 1

What is the immediate impact of this research?
A press release (Taysha Receives Orphan Drug Designation from the European Commission for TSHA-102 for the Treatment of Rett Syndrome | Taysha Gene Therapies (tayshagtx.com) from Taysha Gene Therapies, indicated that the company expected to file an application for an Investigational New Drug (IND) with the US Food and Drug Administration (FDA) in the second half of 2021 and expects to start a Phase 1/2 clinical trial early in 2022. Initial indications are to recruit patients who are older than 18 years and to include younger patients in additional trials.
This is incredible news for the Rett community worldwide particularly as the company has received a special designation for the product, TSHA-102, from the US and European Commission to be considered as a medicine developed to treat patients with rare disease. We will look forward expectantly to the results of the planned Phase 1/2 trial.
Eric Gowans, Adelaide ([email protected]) with extensive editorial input from Mary-Anne Rome and additional help from Karen Rodda, Tony Cagliuso and Gary Grocott.
13/10/2021